

Wir haben uns mit Hani Habra unterhalten, einem der Co-Autoren des Software-Tools Binner: eine Desktop-Anwendung zur Annotation von Isotopen, Addukten und In-Source-Fragmenten in Untargeted Metabolomics-Daten, produziert durch Elektrospray-Ionisations-Flüssigkeitschromatographie-Massenspektrometrie (kurz ESI-LC/MS)
Tools wie Binner helfen dabei, die Komplexität von Untargeted Metabolomics-Datensätzen stark zu reduzieren. Ohne solche Tools wäre es weitaus schwieriger, aussagekräftige Rückschlüsse aus den Messungen zu ziehen, da relevante Informationenen oft durch Rauschen überlagert werden.
Wie ein Metabolit mehrere Signale erzeugt
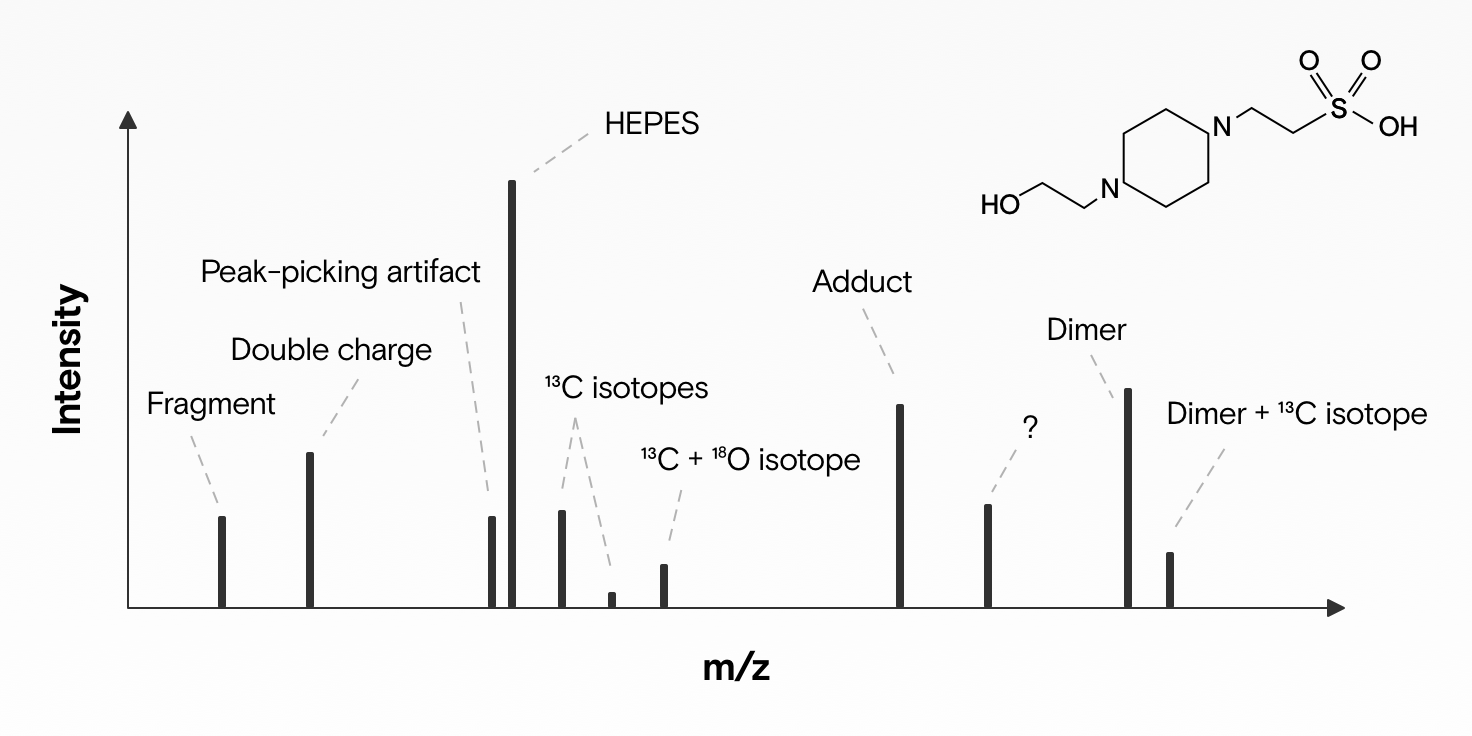
Wenn wir eine biologische Probe (z. B. Blut) mit einem Elektrospray-Ionisations-Massenspektrometer analysieren, dann erzeugt ein einzelner Metabolit (z. B. ein kleines Molekül im Blut) nicht nur ein Signal, sondern mehrere. Dafür gibt es vier Gründe:
- Isotope: In der Regel gibt es von einem Molekül mehrere isotopische Versionen (Isotopologe). Das bedeutet, dass es sich zwar um das gleiche Molekül handelt, eines oder mehrere seiner Atome jedoch eine unterschiedliche Anzahl von Neutronen aufweisen. Ein Beispiel: ~1,1% aller Kohlenstoffatome auf der Erde haben 7 Neutronen und nicht 6. Wenn unser Molekül 1 Kohlenstoffatom hat, dann werden wir wahrscheinlich auch einen zweiten Massenpeak mit ~1 Dalton höherer Masse (M+1) und nur etwa 1.1 % relativer Häufigkeit in unserem Massenspektrum beobachten.
- Addukte: Neue Moleküle entstehen, indem sich zwei oder mehr Moleküle miteinander verbinden. Auch ein Teil der Moleküle, die wir messen wollen, verbinden sich mit anderen zu neuen Molekülen (Addukte). Welche Addukte dabei entstehen können, hängt von mehreren Faktoren ab: Wie die Probe vorbereitet wurde (z. B. welches Lösungsmittel verwendet wird: Aceton oder Ameisensäure), wie die Proben gelagert wurden und ob die Geräte im positiven oder negativen Ionenmodus betrieben werden. Ein einfaches und häufiges Beispiel sind Addukte, die entstehen, wenn ein Natriumatom (Na) oder ein Proton (H+) hinzugefügt wird.
- Fragmente: Wenn Moleküle in einem Massenspektrometer ionisiert werden, können sie energetisch instabil werden und dadurch zerfallen. Normalerweise hat dann eines der entstandenen Fragmente eine neutrale Ladung und ist daher nicht messbar, während andere - jetzt kleinere - geladene Fragment-Ionen bestehen bleiben. Diese Fragmente erscheinen als zusätzliche Signale. Wenn wir wissen, welche Neutralverluste zu erwarten sind, können wir diese Fragmente zusammen mit dem ursprünglichen Molekül gruppieren.
- Multimere: Zwischen zwei oder mehreren Metabolitmonomeren können sich - zusätzlich zu den geladenen Verbindungen - Komplexe bilden, die einen m/z-Wert erzeugen, der typischerweise etwa das 2-3fache des einzelnen Metaboliten beträgt.
Aus einem einzigen Molekül entstehen also viele Signale, was für die Analyse ein erhebliches Hindernis darstellt.
Warum das Splitten von Signalen die Downstream Analysen erschwert
Ein Molekül erzeugt also nicht nur ein, sondern viele Signale. Das macht es uns schwer, genau zu bestimmen, wie viel von einem bestimmten Molekül in der Probe genau vorhanden war, die wir gerade messen.
Wenn wir die absolute Menge eines Metaboliten nicht genau messen können, ergeben sich mehrere Probleme:
- Muster sind schwieriger zu erkennen: Wenn man nach einem Unterschied zwischen zwei Gruppen von Patienten sucht (z. B. zwischen denjenigen, die eine Krankheit haben, und denjenigen, die sie nicht haben), findet man diese Unterschiede viel schwieriger, wenn die relevanten Informationen auf viele Signale verteilt sind. Das gesuchte Muster könnte unter Umständen komplett verschwinden.
- Mehrfachvergleiche verursachen Muster aus Zufall: Je mehr Signale wir untersuchen, desto größer ist das Risiko für nur scheinbar interessante Muster, die in Wirklichkeit durch Zufall entstanden sind (siehe multiple comparisons problem). Je mehr einzelne Vergleiche es gibt, desto stärker steigt das Risiko.
- Features sind redundant: Signale, die von demselben Ion stammen, korrelieren wahrscheinlich stark miteinander. Wenn sich also die Anzahl der interessant erscheinenden Features vervielfacht, wird die Downstream Analyse mühsamer und redundanter.
- Biologische Zusammenhänge bleiben verborgen: Die Korrelationen zwischen redundanten Merkmalen machen es schwieriger, korrelierte Metaboliten zu finden - und damit auch schwieriger, die beteiligten biologischen Prozesse zu erkennen.
- Signale werden falsch identifiziert: Je mehr Signale es gibt, desto eher kommt es zu Fehlern. So kann es passieren, dass wir Signale miteinander in Verbindung bringen, die eigentlich nicht zusammengehören, oder dass wir ein Signal einem anderen Metaboliten zuordnen, als es eigentlich entstammt.
All das macht es für uns erheblich schwerer, aussagekräftige Ergebnisse zu erhalten. Noch dazu könnten wir sogar auf Erkenntnisse stoßen, die schlichtweg falsch sind.
Daher ist es essentiell, so viele der gespaltenen Signale wie möglich korrekt zu einem Signal zu gruppieren. Der Prozess, bei dem bestimmt wird, welche Signale zusammengehören und gemeinsam gruppiert werden sollten, wird Annotation genannt. Wir annotieren Signale indem wir eine Erklärung finden durch welche Änderung (z.B. ein Addukt) sich dieses Signal aus dem ursprünglichen Vorläuferion geformt hat. Und genau dabei hilft uns Binner.
Feature-Annotation vor Binner
Binner ist nicht das erste Tool, das bei der Annotation von Signalen hilft. Seit fast 10 Jahren ist das R-Tool CAMERA sehr verbreitet, und es gibt noch viele andere Tools.
Vor der Entwicklung von Binner gab es fünf wesentliche Techniken, mit Hilfe derer Annotations-Software bestimmt, welche Signale zusammengehören:
- Gruppierung nach Retentionszeit/Thresholding: Die Retentionszeiten der verknüpften Merkmale müssen sehr ähnlich sein - das bedeutet, die Merkmale koeluieren. Im Normalfall stellt das Tool eine bestimmte Toleranz für Retentionszeit-Gruppen ein, und sobald der Unterschied zwischen zwei Signalen größer ist als diese Toleranz, erstellt das Tool eine neue Gruppe.
- Korrelation: Merkmale, die vom gleichen Metaboliten stammen, korrelieren meistens miteinander. Daher berechnet man die Korrelationen aller Merkmale in einer Messung, und diejenigen Merkmale mit höherer paarweiser Korrelation gehören mit größerer Wahrscheinlichkeit zusammen.
- Clustering: Nachdem die Merkmale nach Retentionszeit eingeteilt und die paarweisen Korrelationen berechnet wurden, verwenden die Tools oft einen Clustering-Algorithmus, um die Merkmale anhand einer Ähnlichkeitsmetrik (z. B. der Korrelation) zu gruppieren.
- Chromatographische Ähnlichkeit der Peak-Form: Addukte und Fragmente sollten eine ähnliche Peak-Form haben – da sie ja dem gleichen Metaboliten entspringen, der in einem gewissen Konzentrationsverlauf aus der LC Säule kommt.
- Relative Häufigkeit der Addukte: Auf Basis von Erfahrungswerten und den gemessenen Häufigkeiten verschiedener Addukte in vergleichbaren Datensätzen können wir Annahmen darüber treffen, welche Fragmente plausibler sind als andere.
Binner kombiniert mehrere dieser bestehenden Techniken, geht dann aber noch einen Schritt weiter und bietet die Möglichkeit, Visualisierungen zu erstellen.
Wie Binner Signale annotiert
Wie die anderen Tools gruppiert Binner - das Tool, das Hani Habra mitentwickelt hat - automatisch Signale, die vom selben Metaboliten stammen.
Es zielt darauf ab, die Anzahl an Signalen in einem Datensatz stark zu reduzieren - und dabei so viele Verbindungen zwischen Signalen im Datensatz und realen Molekülen wie möglich zu erhalten.
So funktioniert Binner Schritt für Schritt:
- Einteilung nach Retentionszeit: Binner gruppiert Signale anhand von Unterschieden in der Elutionszeit in eigenständige Einheiten, genannt “Bins”.
- Korrelationen berechnen: Als nächstes wird die paarweise Korrelation auf Basis der Meßwerte aller Signale in einem Bin berechnet.
- Isotope nachweisen: Binner findet und gruppiert C13-Isotopenversionen von Signalen. Dabei geht es davon aus, dass die Häufigkeit von Isotopen mit zunehmender Masse sinkt (da C13 naturgemäß seltener vorkommt als C12).
- Hierarchisches Clustering: Als nächstes clustert Binner die Merkmale anhand ihrer Korrelationsvektoren und optimiert die Anzahl an Clustern, um den durchschnittlichen Silhouettenkoeffizient jedes Merkmals zu minimieren.
- Das am häufigsten vorkommende Signal zentrieren: Es verwendet das Signal, das am häufigsten vorkommt, als Basis für die anschließenden Addukt-Hypothesen.
- Hypothesen testen, um Massendifferenzen zu erklären: Binner untersucht dann die häufigsten Ionen - einschließlich der vom Benutzer erstellten Liste von Addukten und neutralen Verlusten/Zuwächsen - um Massendifferenzen zu erklären. Dann übernimmt er die Hypothese, die die maximale Anzahl von Signalen in einer bestimmten Gruppe annotiert.
- Wiederholungen: Am Ende probiert Binner, auch die verbleibenden Ionen zu annotieren.
Wie die Annotations-Datei Binners Annahmen leitet
Wie bei den meisten anderen Tools stellt man Binner eine Annotationsdatei zur Verfügung, die die Ladungsträger, neutralen Zuwächse und neutralen Verluste enthält, die man in seinem Datensatz vermutet.
Im Gegensatz zu anderen Tools beschränkt sich Binner jedoch nicht auf die genaue Adduktformeln, die man in dieser Datei angibt. Es berücksichtigt auch Addukt-Kombinationen von allem, was in der Annotationsdatei definiert ist. Wenn man also einen Ladungsträger von Na (22.982) und einen Neutralzuwachs von Na - H + NaCOOH (89.97) angibt, dann ist es möglich, dass Binner Merkmale als "M+Na + Na - H + NaCOOH" annotiert (was zu M+2Na-H+NaCOOH vereinfacht werden kann).
Neben der automatisierten Annotationen von Signalen bietet Binner jedoch noch mehr.
Annotationen transparent werden lassen
Binner bietet auch Visualisierungen seiner Annotationen. So kann man nachvollziehen, warum Merkmale auf eine bestimmte Art und Weise gruppiert und beschriftet wurden, wodurch man den Annotationen leichter vertrauen kann.
Binner zeigt auch Informationen an, die man verwenden kann, um neue, bisher unbekannte Addukte zu finden und zu definieren, um sie der Annotationsdatei hinzuzufügen. Dadurch wird die Anzahl der Merkmale, die annotiert werden können, deutlich erhöht.
Schauen wir uns einmal an, wie das funktioniert.
Visualisierung von Signalkorrelationen und Annotationen
Für jeden Cluster, den Binner findet, zeigt es die entsprechenden
- Annotationen, auf die es sich festgelegt hat, sowie die Berechnungen der neutralen Verluste und der Addukte, die zu dieser Annotation geführt haben;
- Korrelationen zwischen den einzelnen Merkmalen des Clusters, sowohl als Werte als auch als Heatmap.

Wenn man die Begründung für jede Annotation kennt, lässt sich auch problemlos eine Plausibilitätsprüfung durchführen. Sieht man zum Beispiel, dass mehrere Addukte so beschriftet wurden: M+COOH+NaCOOH, M+COOH+2NaCOOH, M+Cl+3NaCOOH, M+Cl+4NaCOOH, M+Cl+5NaCOOH, dann weiß man - weil jeder "Schritt" nachvollziehbar ist - dass selbst das sehr komplexe Addukt M+Cl+5NaCOOH vermutlich eine sinnvolle Annotation ist.
Wenn aber "2, 3 und 4" im Cluster fehlen würden, dann ist eine komplexe Addukt Annotation wie M+Cl+5NaCOOH wahrscheinlich nicht korrekt.
Binner liefert auch Hinweise, die dabei helfen, komplexe neue Addukte zu finden. Diesen Prozess nennt Hani "Deep Annotation".
Deep Annotation: Die Suche nach neuen, komplexen Addukten
Auch nach einer gründlichen automatisierten Annotation bleiben viele Merkmale nicht annotiert.
Tatsächlich ist bisher unklar, welche Addukte und Fragment-Ionen aufgrund von In-Source-Ereignissen entstehen können. Daher sind die vorhandenen Annotationsdateien unvollständig - sie enthalten nicht annähernd alle Addukte und Neutralverluste, die auftreten können.
Deep Annotation hilft dabei, bisher unbekannte Addukte zu entdecken, die man gegebenenfalls zu seiner Annotationsdatei hinzufügen möchte:
- Binner sammelt die Massendifferenzen zwischen allen unannotierten Ionen im Datensatz.
- Es berechnet die Häufigkeit der einzelnen Massendifferenzen und markiert die, die am häufigsten vorkommen.
- Anschließend schlägt es vor, welche Addukte diese häufigen Massendifferenzen erklären könnten.
Wenn man die häufigsten Differenzen angezeigt bekommt, erkennt man nicht nur, welche Addukte man seiner Annotationsdatei hinzufügen sollte; man kann auch erklären, wie sich diese - oft komplexen - Addukte bilden könnten. Hier ein Beispiel:
Eine häufige Massendifferenz ist 43,9639 und wird als +2Na-2H annotiert; eine weitere häufige Massendifferenz ist 67,9877, die +NaCOOH entspricht. Zusammen könnten diese eine gemeinsame, aber bisher unbekannte Massendifferenz von 111,952 erklären, die +2Na-2H+NaCOOH entspricht. Diese Information kann man dann in die Annotationsdatei mit aufnehmen.
Die Visualisierung von häufigen Massendifferenzen kann dabei helfen, seine Annotationsdatei sinnvoll zu erweitern - was wiederum Binner dabei hilft, noch mehr bisher unannotierte Signale zu annotieren.
Probiere Binner direkt aus
Du kannst Binner hier direkt herunterladen und installieren: https://binner.med.umich.edu/.
Das Team hinter Binner
Binner wurde von William Duren und Janis Wigginton entwickelt; das Projekt wurde von Dr. Maureen Kachman (Analytische Chemikerin und Geschäftsführerin des Metabolomics Core) und Dr. Alla Karnovsky (Associate Professor an der Computational Medicine & Bioinformatik an der University of Michigan) und Hanis PhD-Advisor geleitet, mit umfangreichen statistischen Mitwirkungen von Dr. George Michailidis.
Bist du gerade dabei, deinen Metabolomics Workflow zu optimieren?
Unser Machine Learning Team kennt sich mit Metabolomics-Workflows bestens aus. Wenn du dir unsicher bist, wie du deinen Forschungscode in zuverlässige Produktionsanwendungen umwandeln kannst, melde dich gerne bei uns.